Science
Research Team Introduces New QM/MM Design Principle for Simulations
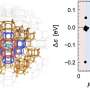
A research team has unveiled a significant advancement in the field of computational chemistry by proposing a new design principle for **QM/MM** (quantum mechanics/molecular mechanics) simulations. This innovative approach allows for the objective and automatic determination of the quantum-mechanical region based on **electronic-state changes**, addressing a crucial challenge faced in multiscale molecular simulations.
The development was detailed in a recent publication in the **Journal of Computational Chemistry**, marking a potential shift in how researchers approach complex molecular systems. By leveraging electronic-state responses, the team aims to enhance the accuracy and efficiency of simulations that are essential in various scientific fields, including materials science and drug design.
Addressing Long-Standing Challenges
For years, the scientific community has grappled with the limitations of traditional **QM/MM** methods, which require manual input and subjective judgment to define the quantum region. The new principle simplifies this process, enabling simulations to adapt dynamically as the electronic states of the system change. This adaptability not only streamlines research workflows but also promises to improve the reliability of simulation results.
The team’s work is particularly relevant as researchers increasingly rely on multiscale simulations to explore complex chemical systems. Enhanced accuracy in these simulations can lead to more effective materials and drug development, ultimately contributing to advancements in technology and healthcare.
Implications for Future Research
The introduction of this design principle could pave the way for broader applications across multiple disciplines. By automating the quantum region determination, future studies will save time and reduce the likelihood of human error, allowing scientists to focus on interpreting results and drawing meaningful conclusions.
As the research community begins to adopt this new methodology, it is expected to stimulate further innovations in simulation techniques. The team behind this development is optimistic that their findings will encourage more researchers to explore the potential of **QM/MM** simulations, potentially transforming the landscape of computational chemistry.
In summary, the proposed design principle presents a promising advancement in the field, with the potential to enhance the quality of multiscale molecular simulations. As this research gains traction, it could significantly impact how scientists approach complex molecular challenges, leading to new breakthroughs in various scientific domains.

Moyes Faces Referee Controversy Ahead of Everton vs Chelsea Clash

Man Hospitalised After Assault Outside Shop in Sandwell

Fred MacAulay and Ally McCoist Reunite for Special World Cup Show

NoOnes Targets $1 Trillion Gift Card Market to Enhance Crypto Access

Beloved Actor Matt Clark Passes Away at 89, Fans Mourn Loss

Discover How Milk Thistle Supplements Enhance Liver Health

Irish Community Care Celebrates 60 Years of Support in Liverpool

New Affordable Housing Development Planned for Crawley Site

Millions of Parking Tickets Issued Amid Calls for Reform

Claire Tomlinson Bids Farewell to Sky Sports After 27 Years

Iconic 90s TV Show House Hits Market for £1.1 Million

Tributes Flow for Kerry Gentle, Beloved RNLI Volunteer and Artist
Nathan Cleary’s Family Celebrates Engagement Amid Romance Rumors

Milk Bank Urges Mothers to Donate for Premature Babies’ Health

Shoppers Flock to Discounted Neck Pillow on Amazon for Travel Comfort

Alessia Russo Signs Long-Term Deal with Arsenal Ahead of WSL Season

Nuneaton Town FC Advances Plans for New Stadium in Stockingford

Museums Body Critiques EHRC Proposals on Gender Facilities
-
 Lifestyle6 months ago
Lifestyle6 months agoClaire Tomlinson Bids Farewell to Sky Sports After 27 Years
-
 Entertainment9 months ago
Entertainment9 months agoIconic 90s TV Show House Hits Market for £1.1 Million
-
 Lifestyle6 months ago
Lifestyle6 months agoTributes Flow for Kerry Gentle, Beloved RNLI Volunteer and Artist
-

 Sports11 months ago
Sports11 months agoNathan Cleary’s Family Celebrates Engagement Amid Romance Rumors
-
 Lifestyle11 months ago
Lifestyle11 months agoMilk Bank Urges Mothers to Donate for Premature Babies’ Health
-
 Lifestyle11 months ago
Lifestyle11 months agoShoppers Flock to Discounted Neck Pillow on Amazon for Travel Comfort
-
 Sports10 months ago
Sports10 months agoAlessia Russo Signs Long-Term Deal with Arsenal Ahead of WSL Season
-
 Sports8 months ago
Sports8 months agoNuneaton Town FC Advances Plans for New Stadium in Stockingford
-
 Politics11 months ago
Politics11 months agoMuseums Body Critiques EHRC Proposals on Gender Facilities
-

 Lifestyle11 months ago
Lifestyle11 months agoExploring England’s Cathedrals: A Journey Through History and Architecture
-

 Business11 months ago
Business11 months agoTrump Visits Europe: Business, Politics, or Leisure?
-

 Lifestyle11 months ago
Lifestyle11 months agoJapanese Teen Sorato Shimizu Breaks U18 100m Record in 10 Seconds